
先天性免疫異常症Inborn errors of immunity, IEI
先天性免疫異常症
山下基 Motoi Yamashita
1-1.先天性免疫異常症の国際分類
20世紀中頃から先天性・遺伝性に免疫系の脆弱性をきたす原発性免疫不全症の疾患概念が徐々に確立してきた。1971年に16の疾患が記載された原発性免疫不全症の分類がWHOから発表された。その後、WHOにおいて原発性免疫不全症を分類し、疾患リストを作成する委員会が組織され、やがて国際免疫学会連合(International Union of Immunological Societies: IUIS)によってPrimary Immunodeficiencies Committee(PID委員会)として運営されるようになった。1990年代から遺伝学的解析技術の進歩と原発性免疫不全症の遺伝学的背景の解明が進み、年々新たな疾患(原因遺伝子)が報告されるようになった。1999年からはIUISのPID委員会によって原発性免疫不全症の国際分類がおよそ2年毎に発表されるようになった。原発性免疫不全症の国際分類には、新たに発見された原発性免疫不全症も含めてすべての原発性免疫不全症が記載され、世界中の臨床医・研究者が参照できるようになった。日本からのPID委員会の委員として野々山恵章先生のあとを森尾友宏先生が継いで、2017年版以降の原発性免疫不全症国際分類の作成を担っている(1-7)。2010年代後半以降、原発性免疫不全症は免疫不全以外のさまざまな免疫異常を含む広い免疫異常の疾患概念として捉えられるようになり、先天性免疫異常症(Inborn errors of immunity, IEI)と呼ばれるようになった。この新しい名称を広く認知させたのも、IUISの国際分類を担うPID委員会がIEI委員会へと名を変え、国際分類にもその名を冠したことが大きな役割を果たしている(3, 4)。2023年1月現在の最新版のIUISによる先天性免疫異常症の国際分類は10の大分類に分かれ485の疾患が含まれている(6)。
1-2.新規先天性免疫異常症の発見:IKAROS異常症とAIOLOS異常症
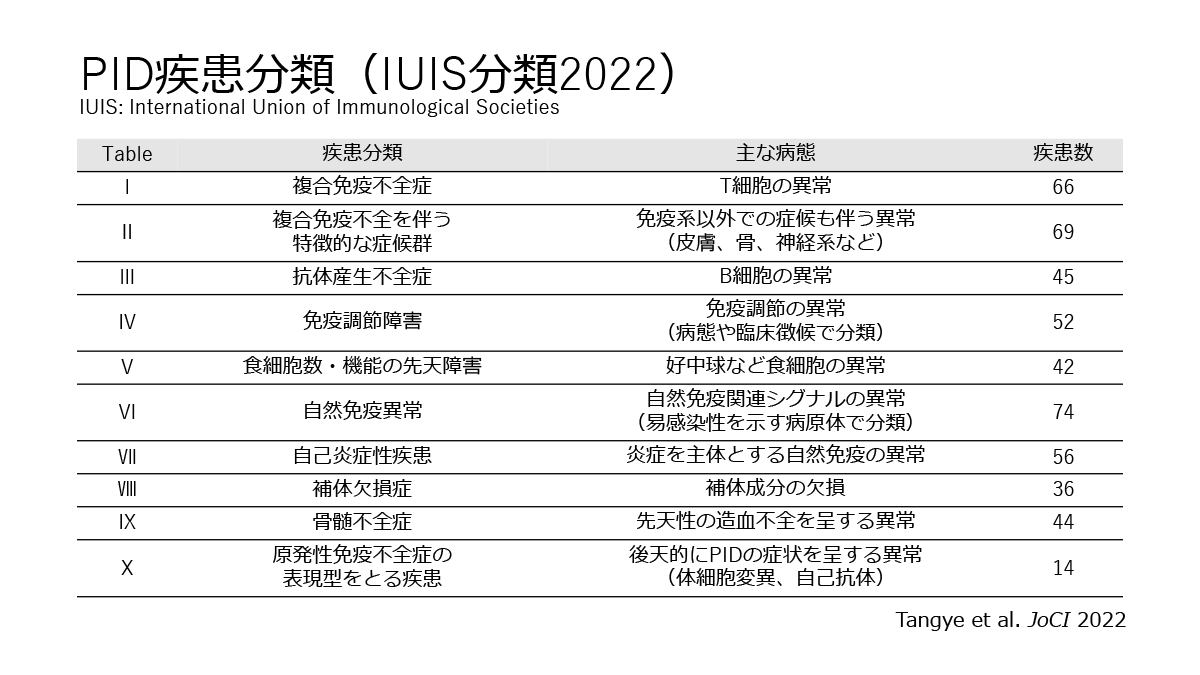
IKAROS Zinc Finger (IKZF)ファミリー転写因子はIKAROSやAIOLOSなど5つの分子が属し主に血球分化を制御する転写因子として知られている。2012年に汎血球減少を呈した乳児例にIKAROSのミスセンスバリアントが同定され報告され、IKAROSの異常が先天性免疫異常症の原因になりうる可能性が示された。その後、2016年、2017年にB細胞欠損症やCVIDの患者コホートからIKAROSのヘテロ接合性バリアントが報告されIKAROS異常症(機能喪失)の疾患概念が確立された(Kuehn HS, et al. N Engl j Med 2016, 8)。特に本邦から星野らによって報告された報告は、経時的に変化する免疫異常や、IKAROS異常症の中でも特異な免疫不全(複合免疫不全症)を示すN159Sミスセンスバリアントを初めて報告しており、IKAROS異常症の研究のなかでも特に重要なものに位置づけられている(8, Boutboul D, et al. J Clin Invest 2018)。IKAROS異常症はその後世界中から報告が相次ぎ、機能喪失型のバリアントだけでなく機能獲得型のバリアントも疾患原性となることが報告されるなど、IKAROS異常症の疾患概念は広がっている(Hoshino A, et al. Sci Immunol 2022)。
さらにIKZFファミリー分子のAIOLOSもB細胞欠損症や抗体産生不全症、複合免疫不全症の原因になることを森尾友宏先生らの国際共同研究グループは明らかにした(9, 10)。特に森尾友宏先生らが主導した研究では、B細胞欠損症、成人期発症のB細胞性リンパ腫、EBウイルスに対する易感染性を特徴とする患者(1家系)に同定されたAIOLOSのミスセンスバリアントは、AIOLOSの結合配列に対する結合能を失う機能喪失型バリアントでありながら異常配列への結合が強固になるネオモルフィックな性質を持つことがわかった。さらにこのミスセンスバリアントを再現したマウスモデルを用いて、初期B細胞分化障害をきたすこと、二量体形成を介したIKAROSの機能障害がその分子病態の一部であることを解明した。先天性免疫異常症の分子病態において、異常をきたした分子自身の機能障害でなく他の分子の機能障害が病態となることは今まで知られておらず、先天性免疫異常症の分子病態の新たな一面を明らかにしたことが注目された(11-13)。
1-3.新規遺伝子治療
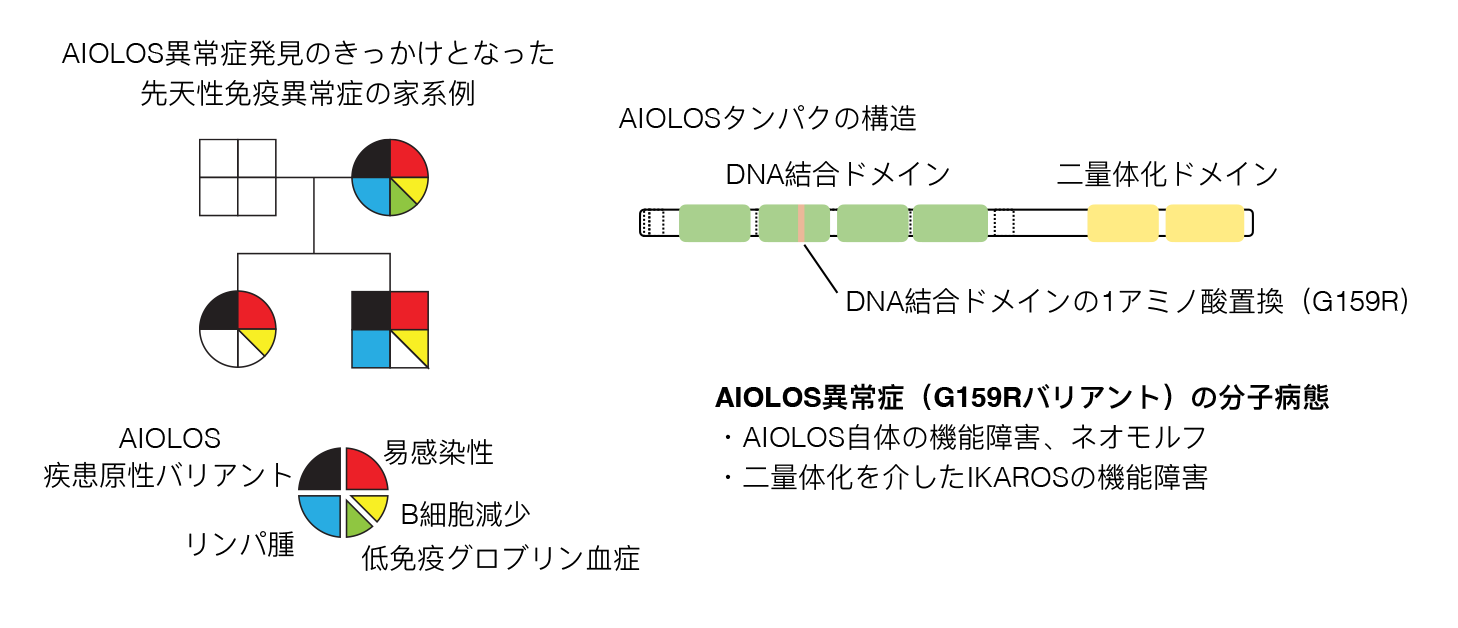
多くの先天性免疫異常症の治療は感染症に対する予防・治療や免疫抑制療法など対症療法であることがほとんどである。根治的療法として骨髄移植などの造血細胞移植が有用であると知られている疾患も一部存在する。先天性免疫異常症の多くは単一遺伝子の異常に起因する疾患であることから、遺伝子治療が根本的な治療法になりうることは初期から期待されていた。特に、造血細胞移植を要するような重症の先天性免疫異常症に対して、造血細胞移植に関わる多くの問題(ドナーの不在、レシピエントの残存する免疫によるグラフトの拒絶、GVHDなどの移植関連合併症)を回避しうる遺伝子治療の開発は必須の課題であった。1990年代からレトロウイルスやレンチウイルスベクターを用いて正常な遺伝子(cDNA)を患者ゲノムに付加的に導入する「gene addition型」の遺伝子治療がいくつかの先天性免疫異常症に対して臨床応用され始めた。一定の疾患では効果をあげる一方で、ウイルスベクターの使用に伴うがん遺伝子の異常活性化とそれに伴う遺伝子導入細胞の癌化が問題となっていた。一方で、患者ゲノム中の疾患原性バリアントを直接修正する「gene correction型」の遺伝子編集治療が2010年代後半以降、CRISPR/Cas9の発見によって現実的となっていった。ただ、CRISPR/Cas9の直接的な遺伝子治療への応用はオフターゲットDNA二重鎖切断後の修復過程で起きる変異導入や染色体構造異常の出現などの副作用が懸念されていた。2024年現在、森尾友宏先生らのグループは先天性免疫異常症に対するニッカーゼタイプのCas9変異体を用いた遺伝子修正治療法の開発に取り組んでいる。この治療法(NICER法:multiple nicks induced by Cas9 nickase and a homologous chromosome as an endogenous repair template)は、常染色体顕性遺伝形式を示す単一遺伝子疾患に対して、疾患原性バリアントを有するアリル特異的なニック(DNA単鎖切断)と、健常アリルにも導入される複数のニックを組み合わせることによって、健常アリルを鋳型とした修正を疾患原性バリアントに対して行う方法で、修復の鋳型として外来DNAを必要としない点や、DNA二重鎖切断を伴わず、オフターゲットの変異導入が非常に少ない利点が知られている(Tomita A, et al Nat Commun 2023)。
- 参考文献
-
- Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018;38(1):96-128.
- Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2018;38(1):129-43.
- Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24-64.
- Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol. 2020;40(1):66-81.
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: an Interim Update by the IUIS Committee. J Clin Immunol. 2021;41(3):666-79.
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473-507.
- Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. 2022;42(7):1508-20.
- Hoshino A, Okada S, Yoshida K, Nishida N, Okuno Y, Ueno H, et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J Allergy Clin Immunol. 2017;140(1):223-31.
- Yamashita M, Kuehn HS, Okuyama K, Okada S, Inoue Y, Mitsuiki N, et al. A variant in human AIOLOS impairs adaptive immunity by interfering with IKAROS. Nat Immunol. 2021;22(7):893-903.
- Kuehn HS, Chang J, Yamashita M, Niemela JE, Zou C, Okuyama K, et al. T and B cell abnormalities, pneumocystis pneumonia, and chronic lymphocytic leukemia associated with an AIOLOS defect in patients. J Exp Med. 2021;218(12).
- Yamashita M, Morio T. AIOLOS Variants Causing Immunodeficiency in Human and Mice. Front Immunol. 2022;13:866582.
- Yamashita M, Morio T. Inborn errors of IKAROS and AIOLOS. Curr Opin Immunol. 2021;72:239-48.
- Yamashita M, Inoue K, Okano T, Morio T. Inborn errors of immunity-recent advances in research on the pathogenesis. Inflamm Regen. 2021;41(1):9.
1-4.OAS1
新しい免疫不全症の病態解明:OAS1異常症による乳児期発症肺胞蛋白と自己炎症症候群
井上健斗 Kento Inoue
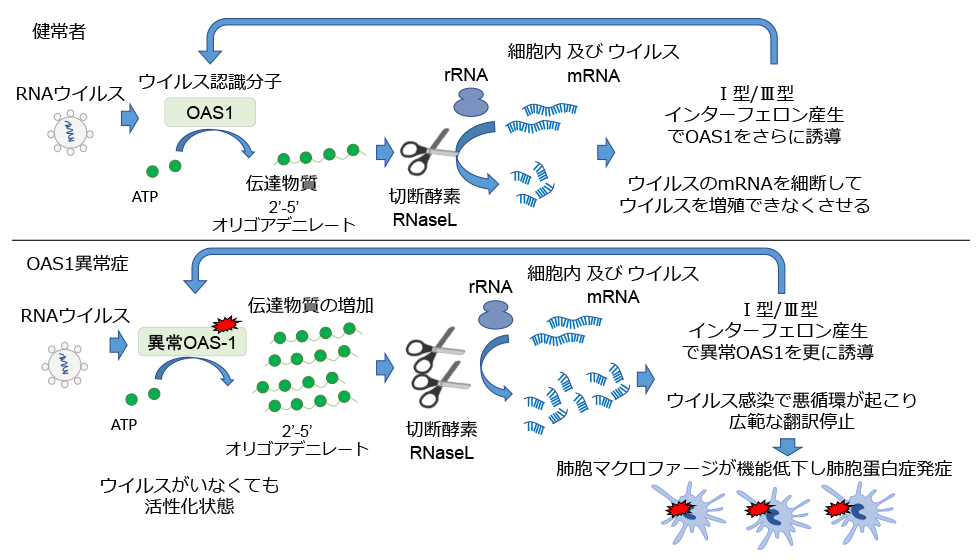
OAS1(2'-5'-Oligoadenylate Synthetase 1)はOASファミリーに分類されるRNAウイルスに対する細胞内センサーとして働く分子である。ウイルス由来の二重鎖RNA(dsRNA)を感知するとATPから2’-5’oligoadenylate(2-5A) を産生する。その2-5Aがsecond messengerとして働き、RNaseL(Ribonuclease-L)を活性化し、ウイルスRNA及び自己のRNAを切断することで広範なタンパク翻訳停止やウイルス複製の抑制、そして1型インターフェロンを誘導し抗ウイルス免疫を活性化させる。1型インターフェロンは更にOAS1の発現を高める。近年OAS1のvariantが新型コロナウイルスの重症化、あるいは逆に重症化防止に関与することがわかり、注目されている。
2018年に北海道大学と共同で本邦におけるウイルス感染を契機とした乳児期発祥の肺胞蛋白症と低免疫グロブリン血症、自己炎症性皮疹を合併する特徴的な症候群を呈する4例の患者の原因遺伝子としてOAS1の特定の変異を同定した 1)。以降、私たちのグループはドイツや米国、オーストラリアなどの大学との国際共同研究によりその病態解明を行ってきた。従来知られていた抗ウイルス免疫分子としてのOAS1の機能だけでは肺胞蛋白症という特徴的な病態が説明困難であり、一方この事実は病態解明の鍵となると考え、肺胞蛋白症の原因となるマクロファージに注目した研究を行った。
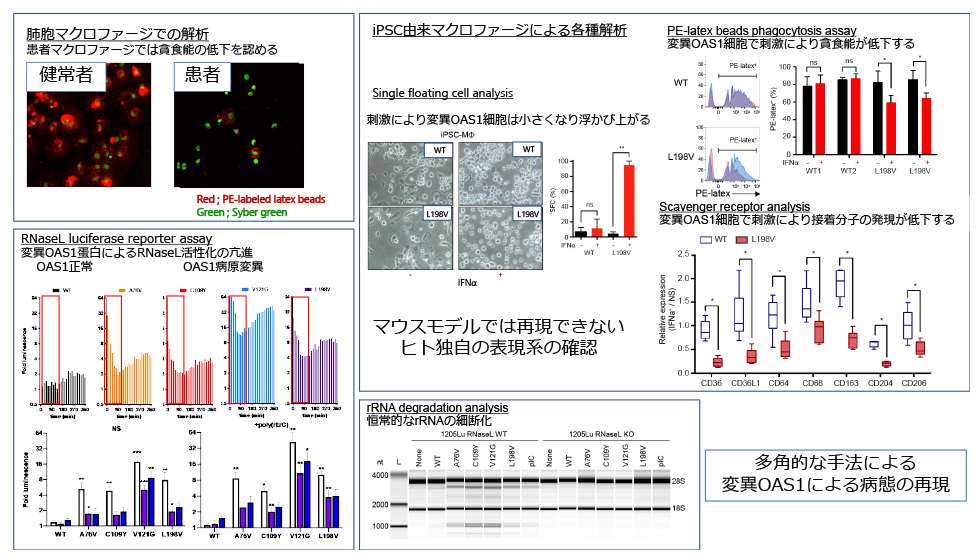
私たちはまず変異OAS1蛋白を用いた患者由来iPS細胞及び変異を導入したiPS細胞を樹立し、マクロファージに分化させ、感染刺激に伴う形態学的な変化や分子発現の変化を観察した。OAS1変異マクロファージはウイルス感染を模したIFNαによる刺激を行うと、形態変化を示し、細胞の接着性を失い、貪食能の低下を示した。また刺激に伴う遺伝子発現を調べると、リボソーマルRNA(rRNA)やトランスファーRNA(tRNA)や、抗原提示、代謝、細胞骨格形成と多岐に渡る様々な遺伝子発現が低下していた。患者のB細胞、単球といった免疫担当細胞ではRNAの分解が顕著であり、このことと併せて、OAS1異常症の患者ではRNaseLの機能亢進によるRNA切断が起こっていることが示唆された。
さらに機能を解析するために、変異OAS1蛋白を作成し、蛍光分子を用いた2-5A産生能を評価したところ、変異OAS1蛋白は刺激に関わらず、2-5A産生能が著しく亢進していることが判明した。変異OAS1を用いた様々な細胞でもRNAの分解が確認され、RNaseLをノックダウンすることで分解が解除されることを確認した。これらのことからOAS1異常症で認める変異OAS1はdsRNA非介在性に恒常的に活性化しており、2-5Aを産生してRNaseLを活性化させ、細胞内RNAの分解、翻訳停止、細胞死を誘導する機能獲得型変異であることが示された。またウイルス感染を模倣するdsRNAによる刺激がOAS1-RNaseLの機能を過剰活性化させてマクロファージの機能低下、細胞死を誘導することが肺胞蛋白症の発症機序であることが示された。
またOAS1異常症の治療法の可能性として、変異iPS細胞での解析によりRNaseL阻害剤として働くクルクミンがOAS1変異細胞の形態変化を是正することが分かった。この研究内容はScience immunology誌に掲載された 2)。
この研究は希少疾患に対する責任遺伝子の同定だけではなく、iPS細胞を作成することにより今まで困難だった病態解明、さらには治療効果の評価を行うことができた点において、意義深い研究である。またOAS1はCOVID-19の重症化・非重症化に寄与する分子であり、今後研究が進み、その活性を適切に制御することができれば、COVID-19のような今後の新興RNAウイルス感染症に対する治療の開発に貢献する可能性がある。
- プレスリリース
- OAS1の機能獲得型バリアントにより重篤な自己炎症・免疫不全症が発症する
https://www.tmd.ac.jp/press-release/20210628-1/ - 文献
-
1) Cho K et al: Heterozygous Mutations in OAS1 Cause Infantile-Onset Pulmonary Alveolar Proteinosis with Hypogammaglobulinemia. Am J Hum Genet 102: 480-486, 2018
2) Magg T et al: Heterozygous OAS1 gain-of-function variants cause an autoinflammatory immunodeficiency. Sci Immunol 6: eabf9564, 2021